ntSynt manuscript published: scalable multi-genome synteny
news ·We are excited to announce the publication of our manuscript, ntSynt: scalable multi-genome synteny using an alignment-free minimizer graph, in BMC Biology.
We are excited to announce the publication of our manuscript, ntSynt: scalable multi-genome synteny using an alignment-free minimizer graph, in BMC Biology.
We are delighted to announce that our scientific manuscript, ntRoot: computational inference of human ancestry at scale from genomic data, is now published in Bioinformatics Advances.
In three new publications, our team and collaborators continue to advance the use of environmental DNA (eDNA) for species monitoring, conservation, and ecosystem health assessment; Efforts that are increasingly vital in our rapidly changing world.
We are excited to share our latest study, Concordance and Dissonance: A Genome-wide Analysis of Self-declared versus Inferred Ancestry in 10,250 Participants from the HostSeq Cohort, now available on bioRxiv.
Our latest publication in Current Protocols presents a streamlined procedure for HLA prediction using streaming long-read sequence alignments.
We are excited to share our latest publication in Protein Science, featuring AMPlify, a machine learning-based tool developed at Birol Lab to identify antimicrobial peptides (AMPs).
Long-read sequencing has revolutionized genome assembly, but achieving high accuracy remains a challenge. Our latest publication in BMC Bioinformatics introduces GoldPolish-Target, a novel tool designed to enhance targeted genome assembly polishing.
High-quality, accessible mitogenome sequences are essential for comparative genomics, phylogenetics, and environmental DNA (eDNA) applications. While genomic sequencing reads are available for diverse species, mitochondrial sequences within these datasets remain largely untapped due to the lack of specialized tools for assembling mitogenomes from short-read libraries.
The 30th Pacific Symposium on Biocomputing (PSB), held in Kona, Hawaii, from January 4-8, 2025, is an international and multidisciplinary conference dedicated to showcasing and discussing cutting-edge research in computational methods for solving problems of biological significance. This year, we are excited to present our latest advancements in ancestry inference using modern DNA sequencing technologies with ntRoot.
The 32nd International Conference on Intelligent Systems for Molecular Biology (ISMB 2024) is held in Montreal, Quebec, Canada from July 12-16, 2024. We are excited to announce that the Birol Lab will be presenting at the conference our latest research advancements, which include: the DNA sequence minimizer-based multi-genome synteny utility ntSynt and the sequence alignment-free ancestry inference technology...
We are excited to announce the publication of “De novo Synthetic Antimicrobial Peptide Design with a Recurrent Neural Network” in the journal Protein Science. In this study, we present AMPd-Up, a novel recurrent neural network tool developed for de novo antimicrobial peptide (AMP) design. AMPd-Up leverages in silico sequence generation to efficiently explore the vast sequence space of...
We are pleased to announce the publication of tAMPer, a cutting-edge deep learning model for predicting peptide toxicity. Published as “Structure-Aware Deep Learning Model for Peptide Toxicity Prediction” in the journal Protein Science, tAMPer integrates amino acid sequence composition with ColabFold-predicted peptide structures through graph and recurrent neural networks. This model aims to expedite antimicrobial peptide (AMP) discovery...
In our Letter to the Editor published in the journal HLA: Immune Response Genetics, we delve into the statistically significant association between the HLA-C*04:01 allele and COVID-19 severity. This association, initially reported by our group in 2020 and 2021, has been replicated in multiple studies, including the extensive CanCOGeN CGEn HostSeq COVID-19 patient cohort (n=9,460). Our...
Our manuscript showcasing aaHash has been published in the Bioinformatics Advances journal. Hashing algorithm aaHash adapts the ntHash algorithm for amino acid sequences and features different hashing levels to represent the biochemical similarities of amino acids. In our tests, aaHash is ∼10X faster than generic string hashing algorithms.
Our manuscript presenting and analyzing the black spruce genome has been published in the G3 journal. Black spruce (Picea mariana [Mill.] B.S.P.) is a dominant conifer species in the North American boreal forest that plays important ecological and economic roles and its genome assembly (18.3 Gbp) and annotation (66,332 protein-coding sequences predicted) are valuable resources for forest genetics research...
Our study on the genomics and transcriptomics characterization of Beauveria bassiana, an entomopathogenic fungus used as a biological agent in agriculture and forestry, was just published in BMC genomics. B. bassiana is of particular interest in regulating the proliferation of the invasive mountain pine beetle (MPB) Dendroctonus ponderosae, a wood-boring insect native to western North America that attacks a...
Our scientific article introducing reference-free long read transcriptome assembler, RNA-Bloom2, was short-listed by Aline Lueckgen editor at Nature Communications, and featured on the Editors’ Highlights under “Biotechnology and methods”.
In our study published in the Environmental DNA journal we present unikseq, a comparative genomics utility that uses words of length k (k-mers) to quickly identify unique regions in genome sequences, which can be used to yield highly specific quantitative real-time polymerase chain reaction (qPCR)-based eDNA assays. In our manuscript, we illustrate its application within an animal...
Our de novo long read genome assembler, GoldRush, and reference-free long read transcriptome assembler, RNA-Bloom2, were published today. Please refer to the GoldRush Nat. Commun. and RNA-Bloom2 Nat. Commun. manuscripts, respectively. The GoldRush long read assembler marks a paradigm shift in long read de novo assembly of large genomes, generating highly contiguous assemblies using an order...
The 27th Annual International Conference on Research in Computational Molecular Biology (RECOMB 2023) was held in Istanbul, Turkey from April 14-19, 2023. The Bioinformatics Technology Lab presented various new algorithms and data structures developed by the group in the past year, including unique+conserved region detection in genome sequences with unikseq, k-mer repeat profiler ntHits and data structure...
After introducing ntLink, our minimizer mapping-based long-read genome assembly scaffolder, as part of the LongStitch pipeline, we added multiple important new features to the tool, which are described in our recently published paper in Current Protocols. These new ntLink features include overlap detection, gap-filling and liftover-based iterations, each of which enable users the generate higher quality final assemblies....
This is our third publication on the NanoSim tool, describing a functionality to simulate nanopore reads for metagenome sequencing experiments. Meta-NanoSim is published in GigaScience and is an integral part of the NanoSim project, freely available from GitHub.
Our manuscript presenting on “Associating Biological Activity and Predicted Structure of Antimicrobial Peptides from Amphibians and Insects” has been published in Antibiotics. Antimicrobial peptides (AMPs) hold great potential as effective alternatives to small molecule antibiotics in the race against antibiotic resistance. In our manuscript we present on the initial discovery of 88 AMPs using our in-house predictors rAMPage...
We just published on btllib: a C++ library with Python bindings for efficient genomic sequence processing in The Journal of Open Source Software (JOSS). The btllib library is implemented in C++, includes a high-level & easy-to-use Python interface, and is freely available on GitHub. The btllib common code library includes specialized DNA/RNA/protein (amino acid) sequence-processing algorithms with efficiency...
Our manuscript presenting ntHash2: recursive spaced seed hashing for nucleotide sequences has been published in Bioinformatics. ntHash2 builds ontop of our popular k-mer and spaced seed nucleotide sequence hashing algorithm, ntHash, with a faster and improved implementation. ntHash2 is freely available on GitHub.
The Research Programmer:
• Develops and implements new algorithms
• Works with the BTL’s large C/C++ code base (create/improve modules, etc.)
• Works on complex biological problems in which analysis of DNA and RNA sequence data requires in-depth evaluation
More info available here
Our manuscript presenting rAMPage: Rapid Antimicrobial Peptide Annotation and Gene Estimation has been published in Antibiotics. Antimicrobial peptides (AMPs) hold great potential as effective alternatives to small molecule antibiotics in the race against antibiotic resistance. In this manuscript, we present rAMPage, a scalable high-throughput bioinformatics pipeline for AMP discovery, and demonstrate its utility in the discovery of 7 active...
The 30th conference on Intelligent Systems for Molecular Biology (ISMB 2022) will be held in a hybrid format (in Madison, Wisconsin, USA and virtually) from July 10-14, 2022. The Bionformatics Technology Lab will be presenting various new algorithms and data structures developed by the group in the past year. We are introducing our new de novo long read assembly...
Our manuscript presenting and analyzing four spruce giga-genomes has been published in The Plant Journal. Spruce trees are widespread in the northern hemisphere, and have great importance both economically and in carbon sequestration. In this manuscript, we assembled and annotated the large and highly repetitive genomes of Sitka spruce, Engelmann spruce, white spruce and interior spruce. Comparative analysis of...
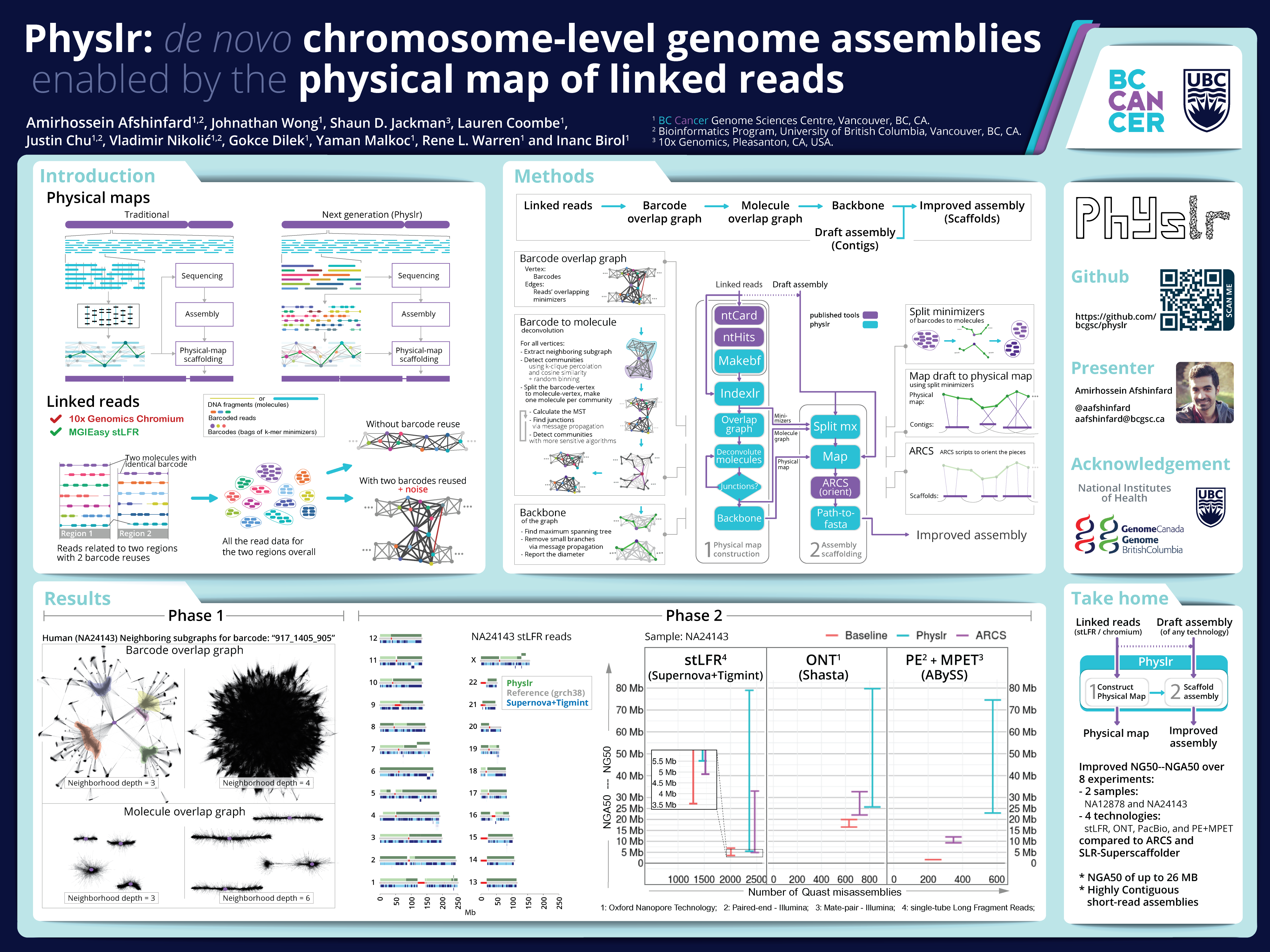
Our manuscript, published in the peer-reviewed journal DNA, presents Physlr, a tool that leverages long-range information provided by linked read sequencing technologies to construct next-generation physical maps. These maps have many potential applications in genome assembly and analysis, including, but not limited to, scaffolding. In our study, using experimental linked-read datasets from two humans, we used Physlr to construct...
The 30th conference on Intelligent Systems for Molecular Biology (ISMB 2022) is taking place in Madison, WI (USA) July 10-14, 2022 and the BTL group will attend in person to showcase our work on de novo long read genome assembler with linear time complexity, GoldRush, and its key components (golden path algorithm GoldRush-Path, long read scaffolder GoldRush-Link and long...
Our manuscript, published in the peer-reviewed journal Current Protocols, presents a fast, scalable and memory-efficient methodology for targeted error resolution and automated finishing of long-read genome assemblies. The ntEdit+Sealer protocol, available on GitHub, was initially designed to polish (any) draft genome assemblies with short sequencing reads. It is being extended to also work with k-mers sourced from erroneous...
In our peer-reviewed manuscript, just published in the journal G3: Genes, Genomes, Genetics, we present the nuclear and mitochondrial genomes and associated annotations of the forest insect pest Pissodes strobi, commonly known as the spruce weevil or white pine weevil, a major pest of spruce and pine forests in North America. We also describe the genome of an apparent...
In our peer-reviewed manuscript, just published in the journal BMC Genomics, we present a novel and robust attentive deep learning model, named AMPlify. In the research manuscript, we show how AMPlify was used to predict antimicrobial peptides (AMPs) from the Rana [Lithobates] catesbeiana (bullfrog) genome and demonstrate the bioactivity of these AMPs against multiple species of bacteria, including multi-drug...
Our manuscript describing an interactive visualization tool for transcripts in single-cell transcriptomes, RNA-Scoop, was just published in NAR Genomics and Bioinformatics. In the manuscript, we show that RNA-Scoop allows users to examine differential transcript expression across clusters and investigate how usage of specific transcript expression mechanisms varies across cell groups. RNA-Scoop is freely available from GitHub.
Our manuscript describing the newly developed long read correction and scaffolding pipeline, LongStitch, was just published in BMC Bioinformatics. LongStitch incorporates multiple tools developed by our group and runs in up to three stages, which includes initial assembly correction (Tigmint-long), followed by two incremental scaffolding stages (ntLink and ARKS-long). Tigmint-long and ARKS-long are misassembly correction and scaffolding utilities, respectively,...
In our peer-reviewed manuscript, just published in the journal PeerJ, we report on observations linking host HLA alleles with COVID-19 disease severity in a New York cohort, some of which (eg. C04:01 and HLA11:01) are corroborated by different research groups and in different COVID-19 patient cohort transcriptome data.
In our peer-reviewed manuscript, just published in the journal IEEE-TCBB, we report on a novel application, GapPredict, for resolving gaps in genome sequence assemblies. This proof-of concept study demonstrates the practical utility of deep-learning machine learning models for this task.
Our manuscript describing Straglr, a tool for both targeted tandem repeat genotyping and novel expansion detection using long-read sequences, was just published in the peer-reviewed journal Genome Biology.
The joint 29th conference on Intelligent Systems for Molecular Biology and the 20th European Conference on Computational Biology (ISMB/ECCB 2021) meeting is held online July 25-30 and the BTL group is presenting several new bioinformatics technologies and analysis workflow developed by our lab. These include ABySS 2.5 genome assembly, LongStitch genome scaffolding, ntEdit/Sealer genome polishing,
A report describing our SARS-CoV-2 variant analysis and interactive SVG mutation maps freely available to the community for browsing and sharing was just published at F1000research. It was announced earlier at arXiv. The maps report nucleotide changes in over 2.5 million SARS-CoV-2 coronavirus genomes (and their effect on gene products) over time and in different continents and...
Our Letter to the Editor, just published in the peer-reviewed journal Bioinformatics, reports on the HLA profiles derived from the metatranscriptomic RNA-Seq samples of eight COVID-19 patients at the pandemic onset. Our study highlights the central role of HLA in vaccine development and host immunity in the current context, and adds perspective to host susceptibility to SARS-CoV-2, the coronavirus...
Our study describing RNA-Bloom, a utility for reference-free and reference-guided sequence assembly of single-cell transcriptomes, was just published in the peer-reviewed journal Genome Research.
Our manuscript describing miBF, a probabilistic data structure we developed for alignment-free sequence classification tasks, was published in PNAS. Alignment-free methods, including miBF, have applications ranging from transcript expression analysis, metagenome characterization, to de novo assembly to name a few. They are usually faster than alignment-based methods, but often limited in their sensitivity and memory requirements. In the manuscript...
The annual International Conference on Intelligent Systems for Molecular Biology (ISMB 2020) is held online July 13-16 and the BTL group will be there, presenting several new bioinformatics technologies developed by our lab and including Meta-NanoSim, ntJoin, Physlr, RNA-Scoop and RResolver.
This is our second publication on the NanoSim tool, describing a functionality to simulate nanopore reads for transcriptome sequencing experiments. Trans-NanoSim was published in GigaScience and is an integral part of the NanoSim project, freely available from GitHub.
Our study describing the genome sequencing of the Sitka spruce mitochondrial genome was accepted for publication in Genome Biology and Evolution. In the manuscript we present the complete 5.5 Mb genome, one of the largest mitochondrial genome of a gymnosperm, assembled from Oxford Nanopore long reads and describe its complex physical structure.
Our manuscript describing ntJoin, a fast and lightweight reference-guided scaffolder, was published in Bioinformatics. Instead of alignments, ntJoin uses a mapping approach based on a graph data structure generated from ordered minimizer sketches. ntJoin can be used in a variety of different research applications, including improving a draft assembly with a reference-grade genome, a short-read assembly with a draft...
Our study describing FusionBloom, a utility for detecting transcript fusions with utility in cancer diagnostics, was just published in the peer-reviewed journal Bioinformatics.
Two studies reporting on the complete chloroplast genomes and gene annotations of white and Engelmann spruce are now published in the peer-reviewed journal Microbiology Resource Announcements.
Our study describing ntEdit, a fast and scalable technology to polish and ‘haploidize’ genome sequences, was just published in the peer-reviewed journal Bioinformatics.
We introduce ORCA, a Docker image with hundreds of bioinformatics tools/dependencies, simplifying software installs. ORCA was recently published in the peer-reviewed journal Bioinformatics.
Our latest work on antimicrobial peptide (AMP) discovery in bullfrogs was just published in the peer-reviewed journal Scientific Reports The paper describes the AMP discovery bioinformatics pipeline and functional assays to test their efficacy against microbes.
Our study presenting Tigmint, a bioinformatics utility to detect and correct errors in genome assemblies using linked reads is published in the peer-reviewed journal BMC Bioinformatics.
Our latest genome assembly scaffolder, ARKS, was just published in the peer-reviewed journal BMC bioinformatics The paper describes a new read alignment-free methodology that employs kmer-based read mapping and improves assembly runtime. In the paper we present benchmarks on human genome draft assemblies and show how linked reads can improve drafts assembled with the same sequencing data further.
The Research Programmer: •Develops and implements new algorithms •Works with the BTL’s large C/C++ code base (improve existing modules, create new ones, etc.) •Works on complex biological problems in which analysis of sequence data requires in-depth evaluation More info available here
RECOMB 2018 will be taking place in Paris, France, from April 21-24th. RECOMB-Seq is one of its four satellite workshops this year, taking place from April 19-20th, bringing together researchers in computational genomics and bioinformatics to discuss new frontiers in gene sequencing.
{kind=link}